Zespół Turnera - wszystko, co musisz wiedzieć na jego temat


Przejrzane i zatwierdzone przez: lekarz José Gerardo Rosciano Paganelli
Zespół Turnera jest zaburzeniem genetycznym, które wpływa na chromosomy płci. Charakteryzuje się całkowitym lub częściowym brakiem chromosomu X, więc dotyczy tylko kobiet. Jego kariotyp to 45X.
Obecność pojedynczego chromosomu X ma ogromny wpływ na rozwój pierwotnych i wtórnych żeńskich cech płciowych. Dowiedz się więcej na temat tego, czym jest zespół Turnera czytając nasz artykuł!
Zespół Turnera i statystyki
Statystycznie, 50% przypadków zespołu Turnera ma kariotyp 45X, gdzie występuje całkowita utrata drugiego chromosomu. Piętnaście procent ma tylko częściową utratę tego chromosomu. W takich przypadkach może to mieć wpływ na długie lub krótkie ramię chromosomu.
Pozostałe przypadki to mozaiki. Charakteryzują się one obecnością dwóch lub więcej linii komórkowych ze zmianami genetycznymi. W ten sposób osoby cierpiące na zespół Turnera będą miały część komórek genetycznie normalnych (45XX) i niektóre genetycznie zmienione (45X).
Chromosomy płciowe w normalnych warunkach
W normalnych warunkach każdy ma 23 pary chromosomów. Spośród nich 22 odpowiadają bezpłciowym autosomom lub chromosomom, a ostatnia para to para chromosomów płciowych.
Chromosomy płciowe, odziedziczone po ojcu i po matce, określają płeć genetyczną dziecka.
- Obecność dwóch chromosomów X (46XX) określa, że dziecko będzie płci żeńskiej. W ten sposób rozwiną się kobiece pierwotne i wtórne cechy płciowe.
- Obecność chromosomu Y (46XY) określa, że dziecko będzie płci męskiej. W ten sposób prowadzi to do rozwoju męskich pierwotnych i wtórnych cech płciowych.
Zobacz także: Płeć dziecka – sposoby na to, by ją poznać
Co zmienia zespół Turnera?
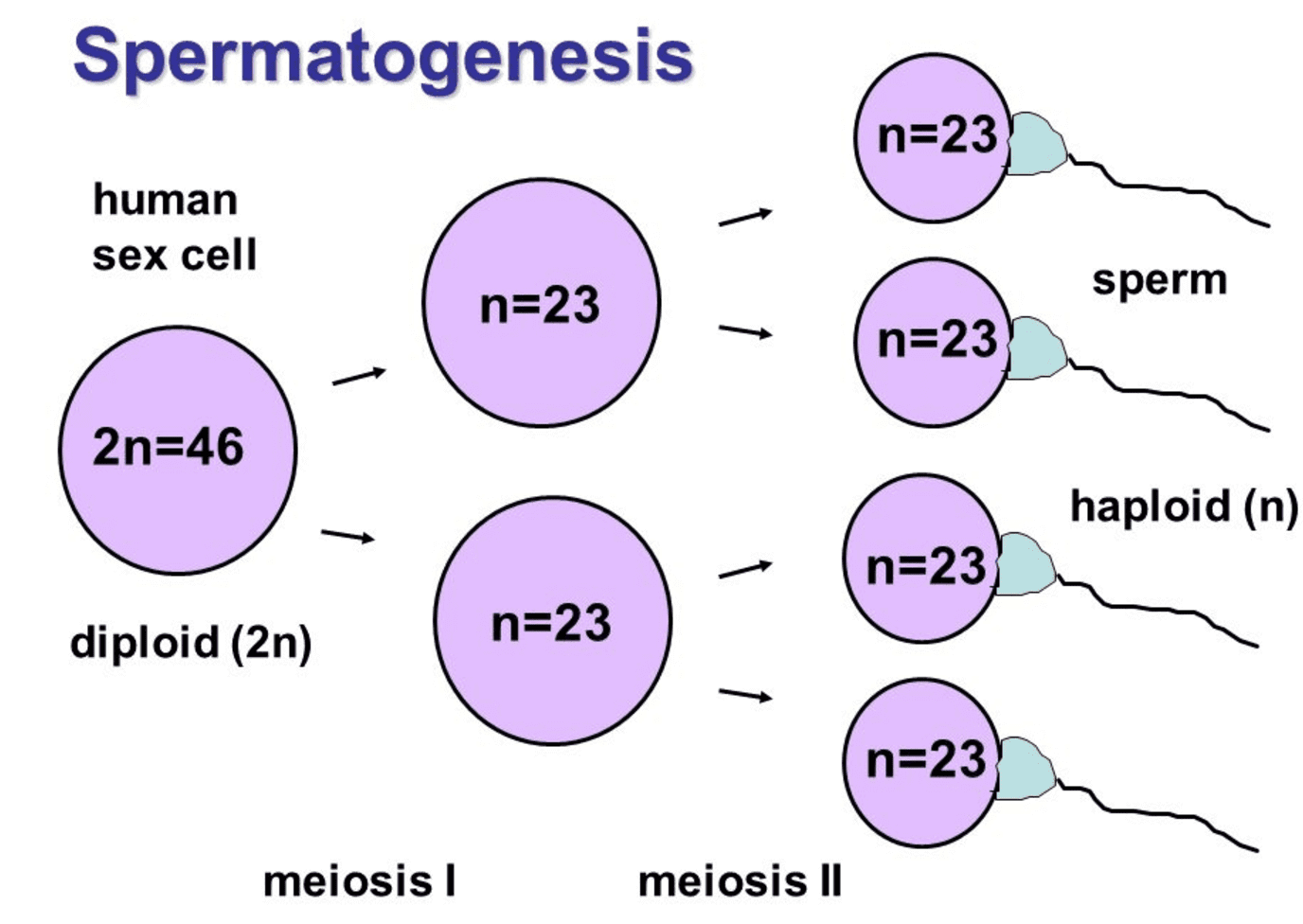
Zanim przejdziemy do wyjaśnienia tej anomalii chromosomowej, ważne jest, aby przypomnieć pewne aspekty rozrodczego podziału komórkowego (gamet).
Proces ten to mejoza i występuje w dwóch fazach, znanych jako mejoza I i mejoza II. Ogólnie rzecz biorąc, celem mejozy jest wytworzenie komórek potomnych z połową informacji genetycznej z pierwotnych komórek.
- Zasadniczo zaczynasz od komórki macierzystej, która zawiera 23 pary chromosomów.
- Po mejozie I powstają dwie komórki potomne, z których każda ma 23 chromosomy.
- Po mejozie II każda z tych dwóch komórek wytworzy dwie inne komórki z 23 chromosomami.
- Dlatego jedna diploidalna komórka macierzysta wytworzy cztery haploidalne komórki potomne.
W jaki sposób komórka traci chromosom?
Dominująca teoria głosi, że utrata tego drugiego chromosomu X lub Y występuje po poczęciu. Oznacza to, że jest to błąd w podziale komórek.
Brak chromosomu Y determinuje rozwój kobiecych cech płciowych. Nawet jeśli oryginalna komórka zawierała chromosomy XY, utrata Y doprowadzi do rozwoju żeńskich organów.
Jakie objawy kliniczne ma zespół Turnera?
Wygląd fizyczny
- Opóźnienie wzrostu podczas dzieciństwa i niski wzrost w wieku dorosłym.
- “Sfinks” – charakterystyczne rysy twarzy, ze spłaszczonym mostem nosowym, nisko osadzonymi oczami i niewielkimi zniekształceniami powiek.
- Krótsza i szersza szyja niż normalnie z bardzo niską linią włosów.
- Liczne znamiona i zmiany pigmentacyjne.

- Szeroka klatka piersiowa i słabo rozwinięte oraz szeroko rozstawione sutki.
- Wtórne cechy płciowe są bardzo słabo rozwinięte, z widocznym płciowym infantylizmem. Niewielkie piersi, bardzo mało włosów łonowych i brak zaokrąglonych bioder. Wszystko to spowodowane jest brakiem estrogenu.
- Wady kości są bardzo powszechne, np. często można zaobserwować odchylenie ramienia na zewnątrz.
Wady wewnętrzne
Niestety, ta przypadłość może również powodować różne zaburzenia wewnętrzne, takie jak:
- Wrodzone wady serca, takie jak koarktacja aorty i inne wady zastawki. Te problemy występują bardzo często.
- Problemy z nerkami mogą pojawić się u znacznej liczby pacjentów, najczęściej występuje nerka podkowiasta.
- Dysgeneza gonad. W tym przypadku jajniki nie są dobrze rozwinięte, a zamiast nich obecne są zespoły gonad. Prowadzi to do hipogonadyzmu hipergonadotropowego, co oznacza, że chociaż wszystkie bodźce potrzebne do produkcji estrogenu są obecne, nie jest on produkowany przez brak jajników.
- Brak miesiączki i bezpłodność. Tylko niewielki procent pacjentów jest płodny. Jest to jedna z najczęstszych cech zespołu Turnera.
Przeczytaj również: Bezpłodność u kobiet – 7 głównych przyczyn
Diagnoza
Zespół Turnera może być trudny do zdiagnozowania. Większość kobiet z tym zaburzeniem nie ma bardzo oczywistych cech: są to łagodniejsze fenotypy, ponieważ w przypadku cięższego uszkodzenia płodu najczęściej dochodzi do naturalnego poronienia.

Około jedna trzecia przypadków zespołu Turnera jest diagnozowana u dzieci w wieku niemowlęcym na podstawie występowania pewnych cech fizycznych i oznak choroby serca. Kolejną część przypadków zdiagnozowano u pacjentów w dzieciństwie, głównie na podstawie niskiego wzrostu.
Ostatnia grupa pacjentów diagnozowana jest w okresie dojrzewania. W takich przypadkach niski wzrost w połączeniu z infantylizmem seksualnym daje pewność, że pacjent cierpi na zespół Turnera.
Leczenie
Hormonalna terapia zastępcza z użyciem estrogenu pomaga w rozwoju cech płciowych, ale nie leczy niepłodności. W dzieciństwie zaleca się leczenie chorych dziewczynek przy użyciu hormonu wzrostu, który pomaga w normalizacji tej cechy.
Czy osoby z zespołem Turnera mogą mieć dzieci?
Tak, niektóre kobiety z zespołem Turnera mogą rodzić dzieci. Niewielki odsetek osób z zespołem Turnera jest płodny i nie ma problemów z zajściem w ciążę.
W przypadku niepłodności, która jest najczęstsza, wymagana jest technika zapłodnienia in vitro przy użyciu pozyskanych wcześniej komórek jajowych. Jajko jest zapładniane przez plemniki in vitro i wszczepiane później pacjentce. Ciąże te wymagają znacznie bardziej rygorystycznego nadzoru lekarza ginekologa niż ciąża normalna.
Wszystkie cytowane źródła zostały gruntownie przeanalizowane przez nasz zespół w celu zapewnienia ich jakości, wiarygodności, aktualności i ważności. Bibliografia tego artykułu została uznana za wiarygodną i dokładną pod względem naukowym lub akademickim.
- Wyss D, DeLozier CD, Daniell J, Engel E. Structural anomalies of the X chromosome: personal observation and review of non-mosaic cases. Clin Genet. 2009; 21(2):145-59.
- Jacobs PA, Betts PR, Cockwell AE, Crolla JA, Mackenzie MJ, Robinson DO, et al. A cytogenetic and molecular reappraisal of a series of patients with Turner’s syndrome. Ann Hum Genet. 1999; 54(Pt 3):209-23.
- Lippe BM. Turner’s syndrome. In: Sperling M. Pediatric endocrinology. 3 ed. Philadelphia: Saunders; 2008.p. 387.
- Chagoyén Méndez, Esther María, Álvarez Montero, José Agustín, & Zúñiga Vaca, Carmen Isabel. (2017). Síndrome de Turner en una adolescente. MEDISAN, 21(6), 720-724. scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1029-30192017000600012&lng=es&tlng=es.
Ten tekst jest oferowany wyłącznie w celach informacyjnych i nie zastępuje konsultacji z profesjonalistą. W przypadku wątpliwości skonsultuj się ze swoim specjalistą.